Unraveling Drug-Induced Hepatotoxicity: Roles of Oxidative Stress, Biomarker Discovery, and Translational Advances
-
Ngabea Murtala
Department of Medicine, Maitama District Hospital, College of Medicine, Baze University, Abuja, Nigeria
Igbayilola Yusuff Dimeji

Department of Human Physiology, College of Medicine and Health Sciences, Baze University, Abuja, Nigeria
| Received 20 Jul, 2025 |
Accepted 02 Sep, 2025 |
Published 03 Sep, 2025 |
The liver is a vital organ that supports numerous bodily functions, including digestion, detoxification, metabolism, immunity, and vitamin storage. It accounts for approximately 2% of an adult’s body weight. The liver is unique because it receives blood from both the portal vein (about 75%) and the hepatic artery (approximately 25%). Hepatotoxicity remains a critical concern in pharmacology and toxicology due to the liver's central role in metabolism, detoxification, and drug biotransformation. A typical adverse effect of several drug classes, drug-induced liver damage (DILI), is usually moderate and goes away quickly upon stopping the drug. Individual susceptibility may be increased by underlying hepatic problems and genetic predisposition. DILI is expected to occur at a rate of 13.9-24.0 cases per 100,000 people worldwide per year. In the United States, DILI is one of the main causes of acute liver failure. The estimated yearly incidence of cases admitted to university hospitals in Korea is 12/100,000 people. Through oxidative stress, mitochondrial dysfunction, inflammation (e.g., TNF-α, IL-6), and metabolic disturbance, xenobiotics, medications, and pollutants can harm the liver. These processes contribute to hepatocellular damage and the advancement of liver disease by causing lipid peroxidation, energy deficit, and fibrosis. Limitations of animal models on account of ethical, translational, and interspecies variability have driven the development of alternative systems. Emerging in vitro platforms, including 3D liver organoids, stem cell-derived hepatocytes, and organ-on-chip systems, provide physiologically relevant systems for hepatotoxicity testing. Augmenting computational tools, such as AI-based models, quantitative structure–activity relationships (QSAR), and physiologically based pharmacokinetic (PBPK) simulations, enhances predictive ability with reduced reliance on animal testing. This review combines up-to-date mechanistic understanding and model developments with an emphasis on integrating molecular data with next-generation strategies to improve hepatotoxicity prediction and human disease relevance.
| Copyright © 2025 Murtala and Dimeji. This is an open-access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. |
INTRODUCTION
The liver is a vital organ responsible for the metabolism of lipids, proteins, and carbohydrates, as well as the degradation of old erythrocytes in collaboration with the spleen. The liver secretes bile for digestion and manufactures plasma proteins that are required, including lipoproteins and factors involved in blood clotting1. Besides metabolism, the liver plays a crucial role in systemic homeostasis regulation through control of immune response, energy generation, and nutrient acquisition2. It also detoxifies drugs and xenobiotics through drug-metabolizing enzymes. Clinically, abnormal liver enzymes typically signify hepatic damage3.
Pharmaceutical and non-pharmaceutical drugs induce drug-induced liver injury (DILI), a significant issue. Risk factors include polypharmacy, liver disease before administration of the precipitant drug, alcohol, genetics, age, and environmental exposure4. The DILI is responsible for about 50% of acute liver failure and causes 5% of hospitalizations. Over 900 compounds are reported to cause DILI, and in severe cases, they result in liver transplant or death in over 75% of affected patients5.
Internationally, the incidence of DILI varies from 2.3/100,000 in the UK and Sweden to 23.8 in China6. Genetic predisposition, alcohol use, and environmental exposures are the substrates for susceptibility7. At the cellular level, hepatotoxicity is driven by mitochondrial damage, impaired fatty acid oxidation, and oxidative stress, the latter resulting from excess production of reactive oxygen species (ROS) and impaired antioxidant defenses8. Sies model distinguishes between eustress oxidative protection and distress oxidative toxicity, the latter being the basis of inflammation and liver damage9.
Natural products like Curcuma, cocoa, L-arginine, walnuts, and Thaumatococcus daniellii possess hepatoprotective effects10-12. Advanced models for example liver-on-a-chip, 3D organoids, iPSC-derived hepatocytes, and multi-omics offer more accurate, human-pertinent data13. These advances must be combined to increase predictability, reduce reliance on animal systems, and guide safer drug development. This study aims to explore the mechanisms underlying drug-induced hepatotoxicity, particularly the role of oxidative stress, evaluate key biomarkers associated with liver injury, and highlight recent translational advances that may enhance the diagnosis and management of drug-induced liver injury (DILI).
Mechanisms of hepatotoxicity
Alterations in liver enzymes: Hepatic enzyme pathway changes, particularly those that handle drug metabolism, are key mechanisms of hepatotoxicity. Leading the process is the cytochrome P450 (CYP450) enzyme family, particularly isoforms like CYP2E1, CYP1A2, and CYP3A4, that metabolize lipophilic drugs to hydrophilic compounds. But oxidative processes yield reactive oxygen species (ROS) and toxic intermediates as well, leading to hepatocyte injury14.
Glutathione S-transferases (GSTs) defend by conjugating glutathione to reactive metabolites. Repeated toxin exposure depletes glutathione, inactivates GST, and increases damage. GST gene polymorphisms also define drug-induced liver injury susceptibility and play a role in liver diseases like NAFLD and hepatocellular carcinoma15. Phase II enzymes and UGTs are also key in detoxification. Inhibition of these can lead to drug clearance being reduced, hence toxic accumulation and liver side effects, particularly with NSAIDs and chemotherapy.
Mitochondrial dysfunction is a second critical mechanism. Electron transport chain (ETC) complex I and III produce ROS and contribute to oxidative stress, promoting apoptosis, necrosis, and fibrosis16. Endogenous antioxidant defenses are typically inadequate, so mitochondrial-targeted antioxidants like MitoQ and idebenone are likely to be therapeutic options17. Combining enzyme profiling with mitochondrial biomarkers can enhance personalized strategies for hepatoprotection.
Dysregulation of lipid and glucose metabolism: Hepatotoxins block liver metabolism, interfering with lipid, carbohydrate, and protein metabolism, ultimately leading to hepatic dysfunctions. As the organizational hub of lipid homeostasis, the liver controls fatty acid oxidation and lipoprotein production.
|
Mitochondrial failure and oxidative stress are caused by carbon tetrachloride and alcohol toxins, leading to lipid peroxidation, abnormal β-oxidation, and hepatic steatosis18. Aflatoxins and certain medications inhibit VLDL secretion, promoting lipid accumulation and NAFLD development.
Carbohydrate metabolism is also impaired. Ethanol and acetaminophen inhibit gluconeogenesis and insulin signaling via mitochondrial injury, increasing the risk of fasting-evoked hepatocellular hypoglycemia19. The BPA and heavy metals also enhance insulin resistance by receptor gene expression disturbance. The ER stress caused by methotrexate and aflatoxins inhibits protein metabolism, leading to hyperammonemia, hypoalbuminemia, and hepatic encephalopathy. Mitochondrial dysfunction increases ROS, promotes apoptosis and inflammation via IL-6 and TNF-α, worsening fibrosis and liver damage2. Targeted interventions for mitochondrial defense, insulin sensitivity, and PPAR signaling through antioxidants and organoid models are key to treating hepatotoxin-induced damage20.
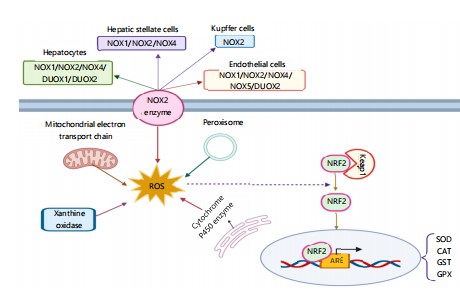
Inflammatory signaling: Oxidative stress, inflammation, and mitochondrial damage are central in hepatocyte necrosis and apoptosis and make the liver highly susceptible to toxic injury. Mitochondrial damage leads to lipid peroxidation, ROS generation, and MPT, all of which are death-promoting. The Nrf2, NF-κB, TNF-α, and IL-6 are the central regulators21 (Fig. 1).
The Nrf2 acts in a protective way by stimulating antioxidant response elements (AREs), which increase detoxification enzymes like glutathione S-transferases (GSTs), superoxide dismutase (SOD), and heme oxygenase-1 (HO-1). Overactivation of oxidative stress or Nrf2 polymorphisms interferes with this process, increasing susceptibility to drug-induced liver injury (DILI) and non-alcoholic steatohepatitis (NASH)22.
On the contrary, NF-κB activation triggers the expression of pro-inflammatory mediators such as TNF-α and IL-6, leading to mitochondrial dysfunction and apoptosis. The TNF-α binding to TNFR1 triggers cytochrome c release and MOMP. The IL-6 promotes liver injury and fibrosis and is a factor in hepatocellular carcinoma. Blockage of these pathways expanding Nrf2 while blocking NF-κB, TNF-α, and IL-6 could yield therapeutic advantages23.
Figure 1 illustrates the source and downstream effects of reactive oxygen species (ROS) in liver disease. Exogenous (for example, alcohol, drugs like acetaminophen (APAP), toxins, viruses, and ultraviolet light) and endogenous (for example, insulin resistance, obesity, dysbiosis of the gut, and increased gut permeability) are the etiologies for ROS production. Metabolism of ethanol and APAP by CYP2E1 generates toxic intermediates (acetaldehyde, NAPQI) that elevate ROS levels. Elevated ROS leads to oxidative damage, including mitochondrial dysfunction (e.g., impaired oxidative phosphorylation, ATP depletion), lipid peroxidation (e.g., MDA, 4-HNE), and oxidized DNA and protein adduct formation, enhancinglipotoxicity. The ROS also triggers inflammation and inflammasome activation, enhancing pro-inflammatory cytokines (IL-1β, TNFα, IL-6) and triggeringpyroptosis and apoptosis/necrosis. Furthermore, altered gut barrier function allows for translocation of microbial products (e.g., LPS, PAMPs/DAMPs), which also enhances hepatic ROS and inflammation. This vicious cycle sustains liver damage and is central to the pathogenesis of liver diseases such as NAFLD, NASH, and drug-induced liver injury.
|
Oxidative stress pathways: Reactive oxygen species (ROS) like Superoxide (O2•–) and Hydrogen Peroxide (H2O2) are natural by-products of mitochondrial respiration and enzyme reactions. Non-replete oxygen reduction in mitochondria generates ROS The Fenton reaction, in which Ferrous Iron (Fe2+) is oxidized to Ferric Iron (Fe3+ ), produces Hydroxyl Radicals (OH•), potent oxidative damage stimulators. Redox cycling re-reduces Fe2+ and continues ROS formation. Nitric oxide (NO•) also combines with O2•– to form peroxynitrite (NOO–), a cytotoxic oxidant linked to lipid, protein, and DNA injury and ferroptosis iron-dependent cell death involving polyunsaturated fatty acid oxidation. While ROS and reactive nitrogen species (RNS) play a role in normal cellular signaling, their excessive production results in oxidative distress. The NO• is safe at physiological concentrations but poisonous in high concentrations25.
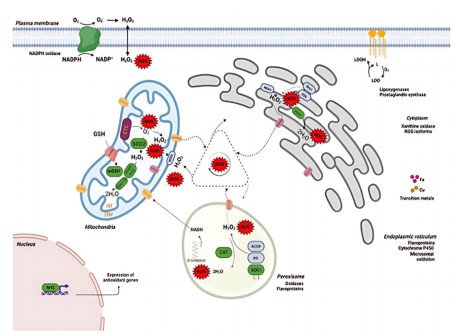
The liver, a detoxification center, is particularly vulnerable due to the high generation of ROS from mitochondrial respiration, peroxisomal oxidases, xanthine oxidase, cytochrome P450s, and NADPH oxidases. Ethanol-induced CYP2E1 is the major ROS producer in hepatocytes. NOX enzymes, especially NOX1-5, are found in Kupffer cells and hepatocytes; noteworthy is that NOX4 generates H2O2 rather than O2•–26 (Fig. 2).
Figure 2 illustrates the generation and detoxification processes of reactive oxygen species (ROS) in hepatocytes. The ROS, including Superoxide (O2•–), Hydrogen Peroxide (H2O2), and Hydroxyl Radicals (•OH), are primarily generated in the mitochondria, endoplasmic reticulum (ER), peroxisomes, and plasma membrane. Electron transport chain complexes of mitochondria, namely Complex I and III, leak electrons
to oxygen to form O2•–, which is dismutated by superoxide dismutase (SOD2) to H2O2. The H2O2 is detoxified by glutathione peroxidase (GPx) and catalase (CAT) or converted to toxic •OH in the presence of transition metals via the Fenton reaction. In the ER, ROS are generated during protein folding and through the activity of enzymes like cytochrome P450 and NADPH oxidase (NOX) isoforms. The cytosol also generates ROS through the activity of xanthine oxidase and nitric oxide synthase (NOS), while lipoxygenases and prostaglandin synthase contribute lipid-derived ROS. Peroxisomes generate H2O2 via fatty acid β-oxidation, which is broken down by catalase. The nucleus functions in response to ROS via the Nrf2 pathway, and this induces antioxidant gene expression for cell protection. Antioxidant mechanisms like GSH (glutathione), SOD (superoxide dismutase), CAT, and peroxiredoxins regulate ROS levels, maintaining redox homeostasis. Dysfunction of these mechanisms leads to oxidative stress, lipid peroxidation, DNA damage, and cell death, and participates in liver diseases like steatosis, fibrosis, and hepatocellular carcinoma.
Antioxidant defense: To counteract oxidative stress, antioxidantsboth enzymatic and non-enzymatic regulate ROS. However, with promising preclinical data, clinical trials have yet to be conducted due to dosing challenges and lacking bioavailability. Endogenous antioxidants include glutathione (GSH), coenzyme Q10, and lipoic acid. GSH, which is produced in the liver, plays a crucial role in detoxification.Coenzyme Q10 protects mitochondrial membranes by participating in electron transport, while lipoic acid recycles vitamins C and E and GSH27.
Biomarkers of hepatotoxicity: Biochemical biomarkers play a very crucial role in diagnosing and following liver disease, as they provide information on hepatic injury and impaired liver function. The liver is under obligation for cardinal processes like bile acid metabolism, protein synthesis, and detoxification, and thus parameters like total bilirubin, plasma bile acids, and plasma proteins are very important for the assessment of liver health. Enzymes like Alanine Aminotransferase (ALT), Aspartate Aminotransferase (AST), glutamate dehydrogenase, and Gamma-Glutamyl Transferase (GGT) are released into the circulation in the event of hepatocyte damage, and they are vital markers for hepatic injury. Besides, disruptions in lipid metabolism characterized by reductions in triglyceride and cholesterol levels signify extreme hepatotoxicity caused by impaired lipoprotein synthesis28.
Moreover, of these biomarkers, indicators linked to oxidative stress have been highlighted for their implication in hepatic inflammation. The GGT, particularly, has been reported to be linked with oxidative stress and inflammatory liver disease, further establishing its diagnostic utility. Oxidative stress plays a central role in hepatotoxicity by inducing lipid peroxidation and mitochondrial injury, further exacerbating liver damage. Because oxidative stress is a hallmark of many liver pathologies, redox balance-associated biomarkers serve equally well for initial diagnosis and follow-up observation of the development of disease. In addition, imbalances in amino acid metabolism have also been regarded as possible markers of liver dysfunction29.
New biomarkers, such as serum arginase I, have shown strong correlations with ALT and AST activity, indicating their potential as early markers of hepatic injury. These new markers are superior to conventional biomarkers because they may identify liver damage earlier, enhancing diagnostic sensitivity. Additionally, advances in proteomics and metabolomics are facilitating the identification of other non-invasive biomarkers that may give a more complete evaluation of liver function and hepatotoxicity30.
In addition, the identification and verification of new non-invasive biomarkers remain a high-priority area of research in hepatotoxicity to enhance diagnostic performance and minimize the reliance on liver biopsies. Because liver biopsies are risky and typically not feasible for regular evaluation, alternative methods of diagnosis are urgently needed. Future research must focus on the integration of multiple biomarkers into predictive models to improve the earlier detection of disease, monitoring of treatment response, and patient outcome in the management of liver disease.
NOVEL BIOMARKERS OF HEPATOTOXICITY
Because of the introduction of system-level approaches towards the evaluation of therapeutics and protein targets for diagnostic purposes (“omics”), there has also been growing interest in targeting proteins, metabolites, DNA/RNA, and their gene products to forecast and obtain possible “toxicity signatures” in drug-induced liver injury (DILI) as well as further our understanding of its pathophysiologic mechanisms31. Metabolomics, on the other hand, offers a high-throughput platform often including mass spectrometry for quantifying these potential liver toxicity markers. However, in current research to find persistent DILI biomarkers, research shows a remarkable lack of predictability concerning the pharmacokinetics and pharmacodynamics of DILI-inducing drugs, with the resultant focus initially being placed upon most commonly implicated ones. It has been suggested by evidence that more than a majority of DILI are due to or are associated with acetaminophen, and this most likely explains why recent biomarker discovery efforts have targeted acetaminophen-induced liver injury. Therefore, much of the clinical and experimental literature that exists on DILI biomarkers has originated from studies of acetaminophen32.
MicroRNA-122 (miRNA-122): Post-translational regulation involves microRNAs, especially miR-122, and is highly specific to hepatocytes, as the liver contains practically 75% of the body’s total miR-122 pool. Both mechanistic and systemic, studies have proved that circulating levels of miR-122 can rise early even preceding the traditional liver injury biomarkers AST and ALT making it a possible diagnostic and risk-stratification marker of DILL. Clinical research indicates that miR-122 levels increase sooner than ALT levels in patients with acute liver injury but remain unchanged in individuals who over-dosed on acetaminophen but did not experience any biochemical liver injury. Increased miR-122 levels have also been associated with worse clinical outcomes, including increased mortality and higher needs for liver support. But how consistent it is, remains to be seen, since miR-122 can be regulated independently of liver damage and exhibits important inter-subject variability. Most data are derived from acetaminophen overdose models, which limits knowledge of idiopathic DILI (IDILI). In addition, many reports are made using non-primate models. Total circulating microRNA profiles are suggested to offer better predictive power than single markers like miR-12233. However, microRNA profiling is a promising approach in the evolving landscape of DILI biomarker identification.
Keratin-18: These cells are mostly epithelial. It is a type-1 intermediate filament that supports the cytoskeleton of cells. Its full-length fragment is released during hepatic cell necrosis, while only its Caspase-cleaved fragment is released during regular planned cell death-apoptosis. The ratio of these two fragments, known as the “apoptosis index” (AI), thereby identifies and may quantify which of the two processes (apoptosis or necrosis) is taking place in a certain index patient with suspicion34.
Later research examining its function in acetaminophen-induced liver damage has revealed a promising mechanistic understanding of the condition. Importantly, in these investigations, it increases before the relative rise in serum transaminases34.
Studies on acetaminophen-induced acute liver injury, both systematic and mechanistic, have contributed much to our current understanding, as has been noted with other possible DILI surrogate signs35. A decisive role in the determination of acute liver injury caused by paracetamol is suggested by the body of information that has so far been gathered from observational and systematic investigations. Validation of this groundbreaking role in other patient populations may be part of the future evidence in this area.
Glutamate dehydrogenase (GLDH): This is mostly found in the liver’s mitochondria, with smaller amounts found in the kidneys and skeletal muscles. It is released into the systemic circulation as a result of hepatocellular damage and the loss of mitochondrial membrane integrity, and it has been demonstrated that its level increases in parallel with an increase in serum ALT after an overdose of paracetamol36. Its constant link with drug-induced hepatocellular injury suggests a major role for it as a novel DILI biomarker, despite a number of confounding factors, such as challenges with varied sensitivities, that have been identified with its test.
High-mobility group box-1 (HMGB-1): High mobility group box 1 (HMGB-1), an associated protein with chromatin, is secreted from necrotic cells and acts on toll-like receptors and receptors for advanced glycation end products (RAGE). A hyperglycated form of HMGB-1 is linked with immune activation, and due to the role of adaptive immunity in DILI, its kinetics can also act both as a biomarker and as a mechanistic marker. HMGB-1-deficient mice undergoing monoclonal antibody therapy, reduced susceptibility to DILI was observed. Some isoforms have also been shown to be of prognostic significance following acetaminophen overdose, such as for mortality and need for liver transplantation. HMGB-1's differential release by necrotic and not apoptotic cells also places its association with necrosis and immune response front and center. A recent prospective study published by Dear et al. in two cohorts (BIOPAR, N = 202, MAPP, N = 985) concluded that the combination of miR-122, HMGB-1, and K18 performed better than ALT alone for the prediction of liver injury risk. Such conclusions herald a new trend towards biomarker-based diagnostics for DILI with HMGB-1 at their core37.
Biomarker validation and clinical utility: Traditional liver injury markers are still crucial for identifying acute hepatic damage from a variety of sources, but their ability to reliably diagnose suspected drug-induced liver injury (DILI) has been limited. Even though interesting DILI biomarkers have been found, their therapeutic use is still developing, especially for idiopathic DILI (IDILI). Recent systematic studies have demonstrated the potential of markers such HMGB-1, miR-122, and keratin-18 (K-18), providing promising diagnostic and prognostic insights. To determine their clinical value, more validation across a range of patient demographics and DILI phenotypes particularly IDILI is essential.
The advent of “omics” technologies as proteomics, metabolomics, and microRNA profiling opened up new possibilities for identifying toxicity profiles unique to DILI. These techniques might reveal more precise and dependable biomarkers38. To strengthen the diagnostic and prognostic power of current biomarkers and facilitate their incorporation into standard clinical practice, more thorough investigation of other candidates and larger, prospectively planned studies are still required.
IN VIVO MODELS FOR STUDYING HEPATOTOXICITY
To evaluate liver injury mechanisms, metabolic disturbances, and therapeutic strategies, in vivo models are essential to study hepatotoxicity. Because of their physiological and genetic resemblance to humans, rodent models, namely, drug-induced liver injury (DILI), Non-Alcoholic Fatty Liver Disease (NAFLD), and chemically induced hepatotoxicity models have been widely employed. Through simulating illness states caused by drugs, environmental toxicants, or diet, the models shed light on hepatotoxic reactions. When extrapolating to human hepatotoxicity, however, species variability in liver biochemistry and immunity must be considered with caution39.
Drugs including acetaminophen, isoniazid, and methotrexate can cause liver damage, usually assessed using rodent models of DILI. For example, acetaminophen liver damage in mice is a well-standardized model that replicates the key clinical findings of human overdose toxicity such as centrilobular necrosis, glutathione depletion, oxidative stress, and mitochondrial dysfunction. Likewise, liver injury models designed by isoniazid model idiosyncratic DILI, which is marked by metabolic changes and immune responses. They enable the investigation of preventive agents that can counteract the hepatotoxin-induced injury, i.e., antioxidants and mitochondrial stabilisers40.
Together with this, the rising prevalence of metabolic liver disease worldwide has created enormous concern in rodent models of NAFLD and DILI. Dietary regimens inducing hepatic steatosis, insulin resistance, and inflammation, such as high-fat, high-fructose, or methionine-choline-deficient (MCD) diets, are widely used to establish NAFLD models. These models effectively replicate human non-alcoholic steatohepatitis (NASH) pathophysiology, such as fibrotic progression to NASH, endoplasmic reticulum (ER) stress, and lipid accumulation. Perhaps most important, these models enable testing for new pharmaceutical compounds that are designed against inflammation, fibrosis, and lipid metabolism41.
Additionally, xenobiotics including aflatoxins, thioacetamide (TAA), and carbon tetrachloride (CCl ) are employed in chemically induced hepatotoxicity models to mimic acute and chronic liver injury. The CCl exposure, for instance, produces reactive metabolites that cause oxidative damage, lipid peroxidation, and hepatic fibrosis and is hence commonly employed in the assessment of the progression of liver injury. Similarly, fibrotic mechanisms and therapeutic interventions on liver regeneration are investigated employing TAA-induced hepatotoxicity models42. Chemical-induced models offer a controlled experimental system to determine hepatotoxin-induced liver damage and discover potential hepatoprotective therapeutics. Genetically engineered animal models and classical rodent models have also allowed identification of distinct molecular mechanisms of hepatotoxicity.
Particular transgenic and knockout mice to the major regulators like Cytochrome P450 Enzymes (CYP450), Tumour Necrosis Factor-Alpha (TNF-α), and Nuclear Factor Wrythroid 2-Related Factor 2 (Nrf2) have shown inflammation, oxidative stress response, and xenobiotic metabolism. The antioxidant defence roles against damage are demonstrated, for example, by the increased sensitivity of Nrf2-null animals to oxidative damage and hepatotoxin-mediated injury. Similarly, knockout mice CYP2E1 provide an insight into how hepatotoxins like ethanol and paracetamol are metabolically activated43.
Limitations: Animal models are widely used in the investigation of hepatotoxicity for the study of liver injury, drug metabolism, and toxic mechanisms. Species differences limit their use for extrapolation to human physiopathology. Rodents, the most widely used models, differ significantly from humans in cytochrome P450 enzyme expression in affecting drug metabolism, toxicity, and bioavailability. Their faster liver regeneration can underestimate chronic injury endpoints compared to human situations. Furthermore, rodent immunity leads to redundant inflammation, decreasing translational value.
High-dose, short-duration animal experimental models are poor models for chronic, low-level human exposures. Metabolic rate and drug clearance variability also make dose extrapolation difficult. Healthy animals are utilized in the majority of studies, whereas numerous human patients harbor intrinsic liver disease (e.g., NAFLD, fibrosis) that affects hepatic metabolism and toxin sensitivity44. Inbred animals lack the genetic heterogeneity found in humans, further lowering predictability. Due to ethical and regulatory constraints, other models are on the rise. In vitro systems, organ-on-chip systems, and 3D liver spheroids offer human-relevant alternatives. While limitations remain, stem cell-derived hepatocytes improve human-specific prediction, redirecting research focus from "translational" animal models to direct human-based systems.
STUDIES OF HEPATOTOXICITY IN PRIMARY CELL CULTURE
Primary hepatocyte cultures are essentially physiologically relevant systems for the investigation of liver-specific functions, drug metabolism, and hepatotoxicity. However, these cultures have severe limitations, including the dedifferentiation of hepatocytes in traditional 2D settings at a high rate, loss of metabolism, and reduced viability with the passage of time. Aside from the above difficulties, there is also a need for continuous tissue sourcing, low proliferative capability, and risks of contamination with non-parenchymal cells. The extracellular matrix (ECM) has been discovered to be crucial in maintaining the structure and function of hepatocytes, and scaffolds such as Matrigel® support keeping patterns of gene expression near native liver tissue. While primary cultures remain a gold standard for hepatotoxicity assays, the disadvantage of non-physiological culture cell density and reduction in CYP450 activity with duration of incubation still prevails. To improve these drawbacks, advanced 3D cultures, co-culture systems, and liver-on-chip technologies increasingly are utilized to preserve liver-specific function and augment the predictive power of in vitro models45.
Organotypic cultures and cellular heterogeneity: While useful, dissociated hepatocyte cultures fail to capture the cellular diversity of the liver or dynamic intercellular interactions. Organotypic cultures attempt to correct this by culturing different types of liver cells, e.g., hepatic stellate cells, Kupffer cells, and liver sinusoidal endothelial cells (LSECs) in more physiologically relevant systems. Co-culture models have been found to exhibit marked improvement in hepatocyte function. For instance, macrophage and hepatocyte co-cultures have delineated significant regeneration and immune response signaling pathways in the liver. Similarly, LSEC-specific features are preserved for hepatocytes co-cultured with LSECs in collagen matrices, and there is increased CYP450 activity. Co-culture with high-oxygen systems has preserved cellular morphology and native gene expression for extended periods of time. One of the most advanced models to date, co-cultures 3T3-J2 fibroblasts, primary human hepatocytes, and LSECs in a Matrigel® overlay, with continued albumin and urea secretion up to three weeks. Such developments mark a milestone in the creation of stable, organotypic platforms for liver research46.
ADVANCES IN THREE-DIMENSIONAL (3D) LIVER CELL CULTURE MODELS
The inherent shortcomings of 2D liver cell culture models have spurred the development of 3D cell culture techniques to more accurately replicate the structural and functional complexity of the liver. 3D liver models are more biomimetic of the hepatic microenvironment in vivo compared to conventional 2D monolayer cultures, which fail to replicate the intricate cell-cell interactions, tissue architecture, and oxygen gradients in vivo. They incorporate some of the main characteristics such as native spatial organization that preserves physiological polarity and increased cell density that echoes the in vivo architecture of hepatic tissue. They also incorporate oxygen zonation that demonstrates the differences between periportal and perivenous areas, and intercellular communication networks that are involved in vital functions of metabolic homeostasis. Additionally, structural components of the hepatic lobule and circulatory system are mimicked in 3D liver models such that dynamic fluidics playing a critical role in the determination of metabolic activity are facilitated. Despite these advantages, there are still limitations with regards to scalability, reproducibility, and long-term culture stability. The majority of 3D models are labor-intensive, lack standardization of methods, and are not readily adapted to high-throughput screening47. Their long-term survival is also in question, which restricts their application in chronic liver disease modeling. The 3D liver culture systems, however, have been extremely useful tools in developmental biology, toxicology, and preclinical drug testing, paving the way for more functionally relevant clinical applications.
IN-SILICO AND AI BASED MODELS
Machine learning and interpretable models: Machine learning (ML) is a new technology for hepatotoxicity research, enabling detection and prediction of the liver toxicity of compounds at a high rate. The conventional methods of the evaluation of hepatotoxicity, such as in vitro and in vivo assays, are generally time-consuming, costly, and ethically demanding. Machine learning (ML) techniques, including ensemble modeling and deep learning, leverage big sets of data, including omics data, histopathology images, and electronic health records, to allow for increased predictive ability and mechanism-based interpretation of DILI.ML integration with computational toxicology allows scientists to better predict liver toxicity biomarkers and identify potential hepatotoxic agents with fewer animal models.
The ML is also responsible for high-throughput screening and analysis of large data in hepatotoxicity research. Support vector machines (SVM), random forests (RF), and neural networks have been applied in predicting toxic and non-toxic molecules from their molecular structure and biological interactions. The models demonstrate the complexity of the relationship between chemical exposure and liver injury through the use of transcriptomic and proteomic data in the prediction of hepatocellular injury. Additionally, natural language processing (NLP) methods applied in biomedical literature and clinical reports enable computerized detection of hepatotoxicity and prediction of risk to support early warning systems in drug development and regulatory decision-making48.
In addition, ML-based methods support precision medicine strategies in hepatotoxicity studies via the individualization of risk assessment according to genetic susceptibility and external exposures of the patients. As an example, predictive models by using input from pharmacogenomic information can foresee patients at risk of DILI and direct therapy interventions in an individualized manner. Moreover, reinforcement learning methods are also being developed to optimize drug dosage regimens with minimal liver toxicity risk. With the advancement of ML, their integration with AI, cloud computing, and blockchain will enhance data security, reproducibility, and transparency of hepatotoxicity research, resulting in safer drug design and improved clinical outcomes.
Physiologically based pharmacokinetic (PBPK) models: Physiologically based pharmacokinetic (PBPK) models are the focal point in hepatotoxicity research since they offer a mechanistic basis for predicting ADME of xenobiotics in the liver. The models use physiological characteristics like tissue partition coefficients, enzyme activity, and hepatic blood flow to simulate drug performance across different populations, i.e., patients with pre-existing liver disease. The integration of in vitro and in vivo data, PBPK models, enhance dose-dependent liver toxicity prediction and helps to identify safe therapeutic windows for drug candidates. In addition, the models extend the risk assessment of occupational and environmental exposure to hepatotoxicants by considering inter-individual variation and species-specific effect49.
Finally, another critical role played by PBPK models in hepatotoxicity studies is their application in drug-drug interaction (DDI) studies, particularly hepatic enzyme inhibition and induction. By incorporating enzyme kinetics and transport-mediated effects, PBPK models are able to simulate how co-administrations are likely to impact hepatic clearance and result in hepatotoxic risk. This aspect is particularly beneficial in determining the part played by cytochrome P450 (CYP) enzymes, which play a central function in drug metabolism and are regularly involved in hepatotoxic. PBPK modeling is equally vital in extrapolating preclinical hepatotoxicity findings to human individuals via the reduction of animal experiments and maximization of the translational value of experimental data. The ability to simulate different patient states, such as phases of liver disease and genetic polymorphisms, enhances hepatotoxicity risk prediction accuracy in diverse populations .
Artificial intelligence: Artificial intelligence is increasingly being applied in all but a few medical subspecialties, with new approaches for disease diagnosis, management, and treatment. Hepatology is not lagging, as AI-driven solutions are being prepared to deal with the growing burden of liver disease, including metabolic dysfunction-associated steatotic liver disease (MASLD) and hepatocellular carcinoma (HCC)–a leading cause of cancer-related deaths worldwide. The growing load of chronic liver diseases needs timely, accurate diagnosis and appropriate treatment approaches, and AI-powered innovation is particularly pertinent to address this need. Besides that, AI has been demonstrated as an efficient tool in predicting patient outcomes and clinical decision-making based on large data. One of the most widespread AI approaches is machine learning (ML), which is a set of algorithmic models that get better at making predictions and diagnostics more precise with time. The ML approaches may be categorised into three major sets:
| • | Supervised learning, where the prediction models are educated with labelled data | |
| • | Unsupervised learning, where the hidden patterns in the unlabeled data are uncovered | |
| • | Reinforcement learning, which allows algorithms to learn incrementally to make better decisions through trial and error |
The AI has been extremely useful across a wide range of applications in hepatology, ranging from the interpretation of structured electronic health records (EHRs) and clinical texts through the application of natural language processing (NLP) techniques to the grading of liver fibrosis severity as well as the identification of early-stage cirrhosis. AI also does better in differentiation of decompensated vs. compensated cirrhosis, grading and detection of portal hypertension, focal liver lesions, e.g., discrimination of HCC from benign liver masses. The AI has likewise enhanced preoperative staging of HCC in patients, monitoring response to treatment, prediction of graft survival in liver transplant recipients. These developments show the promise of AI to enhance the diagnosis and management of liver disease with greater accuracy, efficacy, and reproducibility in the clinical environment.
QUANTITATIVE STRUCTURE AND ACTIVITY RELATIONSHIP
Because they provide a computational framework for predicting the risk potential of chemical compounds from their molecular structure, Quantitative Structure-Activity Relationship (QSAR) models have a significant position in the investigation of hepatotoxicity. These types of models utilize numerical algorithms for formulating correlations among structural features (e.g., functional groups, electrical charges, or molecular descriptors) and documented hepatotoxic impacts in the liver. The QSAR models, through analysis of large sets of known hepatotoxic and non-hepatotoxic compounds, can reliably predict liver toxicity in new or unfamiliar chemicals, with less in vivo experimentation. This is especially useful at early drug development and environmental risk studies, where rapid screening of large numbers of compounds is crucial.
Advances in data integration and machine learning have boosted the predictive power of QSAR models in hepatotoxicity studies. To enhance sensitivity and specificity, contemporary QSAR techniques now include omics-based biomarkers, molecular docking results, and high-throughput screening data. The upgraded models are able to forecast compounds that may result in oxidative stress or inhibit metabolic processes, alongside triggering direct liver injury. To enable the manufacture of safer chemicals and drugs with lower hepatic liability, QSAR models are rapidly becoming indispensable tools in risk assessment regimes as increasingly more regulatory agencies realize the utility of in silico approaches50.
FUTURE PERSPECTIVES
Ethical concerns, cost, and scaling problems still limit conventional hepatotoxicity prediction models, essentially relying on in vivo animal testing and in vitro assays. On the other hand, a rapid, low-cost, and scalable approach to hepatotoxicity assessment is provided by a choice model that incorporates machine learning-based quantitative structure-activity connection. The computer model discussed above supports extrapolation between species and custom projections and is hence better suited to drug safety assessment. Their accuracy, however, is dependent on the quality of training datasets that require further optimization and validation.
Whereas hope of another model persists, management and standardization of verification are still areas of concern. Lack of an accepted standard computer architecture calls for harmonization of rules to create the reproducibility and trustworthiness of models by management agencies across many domains. Future advancements should incorporate hybrid mold strategy that is used to enhance in vitro and in vivo analysis, specifically a multi-tiered liability valuation method. In addition, Explainable Machine Learning Systems (XAI) models development can improve the consistency and trust in machine knowledge prediction and make it easier to manage credibility. Strong interdisciplinary collaboration between toxicologists, computer experts, pharmacists, and regulatory bodies. In addition, integration of omics platforms (generacy, proteomics, and metabolomics) with an artificial intelligence-based PBPK model could improve the validity of hepatotoxicity prediction. Furthermore, there is a need for collaboration among academia, industry, and regulatory agencies to advance computational models to clinically relevant and regulatory-relevant devices.
CONCLUSION
Because of intricate interactions between metabolic derangements and mechanisms of oxidative stress, hepatotoxicity is still a significant problem for toxicology and pharmacology. Generation of reactive oxygen species (ROS), suppression of biotransformation pathways, and dysregulated mitochondrial function are key inductors of liver injury, according to the mounting evidence. Phase I and phase II metabolizing enzymes participating in drug metabolism can be compromised during drug-induced liver injury (DILI), according to in vitro systems and experimental models. This tends to result in the production of hazardous metabolites that worsen cellular injury. Moreover, zonal heterogeneity in oxygen gradients and expression of hepatic enzymes introduces an additional layer of complexity that further compels risk estimation and treatment planning to be even more accurate. In the future, the confluence of AI organ-on-a-chip and multi-omics data holds revolutionary promise for improved hepatotoxic response prediction. Personalized hepatotoxicity risk profiles are being made possible by emerging computational models that combine deep learning with mechanistic understanding of liver metabolism. Emerging research must address the creation of physiologically realistic, human-relevant models that include patient-specific information to move forward in this area. Interpretation of laboratory findings for safer drug development, personalized medicine, and improved regulatory policies on hepatotoxicity screening will involve bridging the experimental systems to actual clinical endpoints.
SIGNIFICANCE STATEMENT
With an emphasis on oxidative stress, important biomarkers, and translational techniques in drug-induced liver damage (DILI), this study investigates the processes of hepatotoxicity. Early identification, prevention, and therapeutic intervention all depend on an understanding of these pathways. This study aids safer medication development by improving the predicted accuracy of DILI models by combining molecular insights with clinical indicators. The results provide a thorough framework for enhancing liver safety assessment in pharmacology by bridging the experimental and clinical domains.
REFERENCES
- Ramatchandirin, B., A. Pearah and L. He, 2023. Regulation of liver glucose and lipid metabolism by transcriptional factors and coactivators. Life, 13.
- Kalra, A., E. Yetiskul, C.J. Wehrle and F. Tuma, 2023. Physiology, Liver. StatPearls Publishing, Treasure Island.
- Bergasa, N.V., 2022. Primary Biliary Cholangitis. In: Clinical Cases in Hepatology, Bergasa, N.V. (Ed.), Springer, London, United Kingdom, ISBN: 978-1-4471-4715-2, pp: 27-84.
- Lee, W.M., 2003. Drug-induced hepatotoxicity. N. Engl. J. Med., 349: 474-485.
- Ostapowicz, G., R.J. Fontana, F.V. Schiødt, A. Larson and T.J. Davern et al., 2002. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med., 137: 947-954.
- Shen, T., Y. Liu, J. Shang, Q. Xie and J. Li et al., 2019. Incidence and etiology of drug-induced liver injury in Mainland China. Gastroenterology, 156: 2230-2241.E11.
- Fisher, K., R. Vuppalanchi and R. Saxena, 2015. Drug-induced liver injury. Arch. Pathol. Lab. Med., 139: 876-887.
- Sies, H., 2015. Oxidative stress: A concept in redox biology and medicine. Redox Biol., 4: 180-183.
- Joshi, A.U., P.S. Minhas, S.A. Liddelow, B. Haileselassie, K.I. Andreasson, G.W. Dorn and D. Mochly-Rosen, 2019. Fragmented mitochondria released from microglia trigger A1 astrocytic response and propagate inflammatory neurodegeneration. Nat. Neurosci., 22: 1635-1648.
- Adeoye, S.W., O. Seun, I.Y. Dimeji, O.E. Ugochukwu and H.L. Jabba, 2025. Hepatoprotective effects of curcumin against dichlorvos-induced toxicity in male Wistar rats. J. Appl. Life Sci. Int., 28: 41-54.
- Igbayilola, Y.D., O.S. Aina, O.O. Ogunkoya, O.D. Williams and F.A. Olaoye, 2022. Oxidative, hepatoprotective and ant-inflammatory responses to perinatal walnut (Juglans regia L.) supplemented diet in offspring of Sprague-Dawley rats. Int. J. Biochem. Physiol., 7.
- Dimeji, I.Y., G.M. Gujja, S.W. Adeoye, L.J. Hamidu, S. Jibrin and Z.M. Baba, 2024. Flavonoid-rich cocoa extract alleviates oxidative stress, inflammation and liver dysfunction induced by high-fat diets in Wistar rats. Res. J. Phytochem., 18: 20-33.
- Russmann, S., G.A. Kullak-Ublick and I. Grattagliano, 2009. Current concepts of mechanisms in drug-induced hepatotoxicity. Curr. Med. Chem., 16: 3041-3053.
- Chanhom, N., S. Wattanapokayakit, N. Satproedprai, S. Suvichapanich and S. Mahasirimongkol et al., 2021. CYP2E1, GSTM1, and GSTT1 genetic polymorphisms and their associations with susceptibility to antituberculosis drug-induced liver injury in Thai tuberculosis patients. Heliyon, 7.
- Webb, M., D.P. Sideris and M. Biddle, 2019. Modulation of mitochondrial dysfunction for treatment of disease. Bioorg. Medic. Chem. Lett., 29: 1270-1277.
- Knockaert, L., A. Berson, C. Ribault, P. Prost and A.A. Fautrel et al., 2012. Carbon tetrachloride-mediated lipid peroxidation induces early mitochondrial alterations in mouse liver. Lab. Invest., 92: 396-410.
- Tan, H.K., E. Yates, K. Lilly and A.D. Dhanda, 2020. Oxidative stress in alcohol-related liver disease. World J. Hepatol., 12: 332-349.
- Guo, K. and T. van den Beucken, 2024. Advances in drug-induced liver injury research: In vitro models, mechanisms, omics and gene modulation techniques. Cell Biosci., 14.
- Arjunan, A., D.K. Sah, Y.D. Jung and J. Song, 2022. Hepatic encephalopathy and melatonin. Antioxidants, 11.
- Aladaileh, Abukhalil, Saghir, Hanieh and Alfwuaires et al., 2019. Galangin activates Nrf2 signaling and attenuates oxidative damage, inflammation, and apoptosis in a rat model of cyclophosphamide-induced hepatotoxicity. Biomolecules, 9.
- Ngo, V. and M.L. Duennwald, 2022. Nrf2 and oxidative stress: A general overview of mechanisms and implications in human disease. Antioxidants, 11.
- Papa, S., C. Bubici, F. Zazzeroni and G. Franzoso, 2009. Mechanisms of liver disease: Cross-talk between the NF-κB and JNK pathways. Biol. Chem., 390: 965-976.
- Jadeja, R.N., R.V. Devkar and S. Nammi, 2017. Oxidative stress in liver diseases: Pathogenesis, prevention, and therapeutics. Oxid. Med. Cell. Longevity, 2017.
- de Almeida, A.J.P.O., J.C.P.L. de Oliveira, L.V. da Silva Pontes, J.F. de Souza Júnior and T.A.F. Gonçalves et al., 2022. ROS: Basic concepts, sources, cellular signaling, and its implications in aging pathways. Oxid. Med. Cell. Longevity, 2022.
- Alengebawy, A., S.T. Abdelkhalek, S.R. Qureshi and M.Q. Wang, 2021. Heavy metals and pesticides toxicity in agricultural soil and plants: Ecological risks and human health implications. Toxics, 9.
- Vermot, A., I. Petit-Härtlein, S.M.E. Smith and F. Fieschi, 2021. NADPH oxidases (NOX): An overview from discovery, molecular mechanisms to physiology and pathology. Antioxidants, 10.
- Mantle, D. and I.P. Hargreaves, 2020. Coenzyme Q10 supplementation in non-alcoholic fatty liver disease: An overview. Gastrointestinal Nurs., 18: 22-27.
- Alves-Bezerra, M. and D.E. Cohen, 2018. Triglyceride metabolism in the liver. Compr. Physiol., 8: 1-22.
- Zhang, Y., L. Zhan, L. Zhang, Q. Shi and L. Li, 2024. Branched-chain amino acids in liver diseases: Complexity and controversy. Nutrients, 16.
- Gîlcă-Blanariu, G.E., D.S. Budur, D.E. Mitrică, E. Gologan and O. Timofte et al., 2023. Advances in noninvasive biomarkers for nonalcoholic fatty liver disease. Metabolites, 13.
- Lagos-Quintana, M., R. Rauhut, A. Yalcin, J. Meyer, W. Lendeckel and T. Tuschl, 2002. Identification of tissue-specific microRNAs from mouse. Curr. Biol., 12: 735-739.
- Antoine, D.J., R.E. Jenkins, J.W. Dear, D.P. Williams and M.R. McGill et al., 2012. RETRACTED: Molecular forms of HMGB1 and keratin-18 as mechanistic biomarkers for mode of cell death and prognosis during clinical acetaminophen hepatotoxicity. J. Hepatol., 56: 1070-1079.
- Kuna, L., I. Bozic, T. Kizivat, K. Bojanic and M. Mrso et al., 2018. Models of drug induced liver injury (DILI)-Current issues and future perspectives. Curr. Drug Metab., 19: 830-838.
- Mohar, I., B.D. Stamper, P.M. Rademacher, C.C. White, S.D. Nelson and T.J. Kavanagh, 2014. Acetaminophen-induced liver damage in mice is associated with gender-specific adduction of peroxiredoxin-6. Redox Biol., 2: 377-387.
- Zhou, Z., J. Qi, Y. Wu, C. Li, W. Bao, X. Lin and A. Zhu, 2023. Nuciferine effectively protects mice against acetaminophen-induced liver injury. Antioxidants, 12.
- Park, J.S., N. Rustamov and Y.S. Roh, 2023. The roles of NFR2-regulated oxidative stress and mitochondrial quality control in chronic liver diseases. Antioxidants, 12.
- Dear, J.W., J.I. Clarke, B. Francis, L. Allen and J. Wraight et al., 2018. Risk stratification after paracetamol overdose using mechanistic biomarkers: Results from two prospective cohort studies. Lancet Gastroenterol. Hepatol., 3: 104-113.
- Pognan, F., M. Beilmann, H.C.M. Boonen, A. Czich and G. Dear et al., 2023. The evolving role of investigative toxicology in the pharmaceutical industry. Nat. Rev. Drug Discovery, 22: 317-335.
- Stine, J.G. and N.P. Chalasani, 2017. Drug hepatotoxicity: Environmental factors. Clin. Liver Dis., 21: 103-113.
- Metushi, I., J. Uetrecht and E. Phillips, 2016. Mechanism of isoniazid-induced hepatotoxicity: Then and now. Br. J. Clin. Pharmacol., 81: 1030-1036.
- Flessa, C.M., N. Nasiri-Ansari, I. Kyrou, B.M. Leca and M. Lianou et al., 2022. Genetic and diet-induced animal models for non-alcoholic fatty liver disease (NAFLD) research. Int. J. Mol. Sci., 23.
- Choi, S.Y., P. Song, J.S. Hwang, Y.K. Lee and M.S. Shin et al., 2024. Cereblon deficiency ameliorates carbon tetrachloride-induced acute hepatotoxicity in HepG2 cells by suppressing MAPK-mediated apoptosis. Front. Immunol., 15.
- Liu, S., J. Liu, Y. Wang, F. Deng and Z. Deng, 2025. Oxidative stress: Signaling pathways, biological functions, and disease. MedComm, 6.
- Lau, J.K.C., X. Zhang and J. Yu, 2017. Animal models of non‐alcoholic fatty liver disease: Current perspectives and recent advances. J. Pathol., 241: 36-44.
- Frojdenfal, S. and A. Zuchowska, 2024. Advanced liver-on-a-chip model for evaluating drug metabolism and hepatotoxicity. Biosensors, 14.
- Ware, B.R., M.J. Durham, C.P. Monckton and S.R. Khetani, 2018. A cell culture platform to maintain long-term phenotype of primary human hepatocytes and endothelial cells. Cell. Mol. Gastroenterol. Hepatol., 5: 187-207.
- Xu, Q., 2021. Human three-dimensional hepatic models: Cell type variety and corresponding applications. Front. Bioeng. Biotechnol., 9.
- Guo, W., J. Liu, F. Dong, M. Song and Z. Li et al., 2023. Review of machine learning and deep learning models for toxicity prediction. Exp. Biol. Med., 248: 1952-1973.
- Shi, Z., L. Huang and H. Wang, 2025. Predictive modeling of pediatric drug-induced liver injury: Dynamic classifier selection with clustering analysis. Digital Health, 11.
- Zhao, M., J. Ma, M. Li, Y. Zhang and B. Jiang et al., 2021. Cytochrome P450 enzymes and drug metabolism in humans. Int. J. Mol. Sci., 22.
How to Cite this paper?
APA-7 Style
Murtala,
N., Dimeji,
I.Y. (2025). Unraveling Drug-Induced Hepatotoxicity: Roles of Oxidative Stress, Biomarker Discovery, and Translational Advances. Trends in Applied Sciences Research, 20(1), 56-69. https://doi.org/10.3923/tasr.2025.56.69
ACS Style
Murtala,
N.; Dimeji,
I.Y. Unraveling Drug-Induced Hepatotoxicity: Roles of Oxidative Stress, Biomarker Discovery, and Translational Advances. Trends Appl. Sci. Res 2025, 20, 56-69. https://doi.org/10.3923/tasr.2025.56.69
AMA Style
Murtala
N, Dimeji
IY. Unraveling Drug-Induced Hepatotoxicity: Roles of Oxidative Stress, Biomarker Discovery, and Translational Advances. Trends in Applied Sciences Research. 2025; 20(1): 56-69. https://doi.org/10.3923/tasr.2025.56.69
Chicago/Turabian Style
Murtala, Ngabea, and Igbayilola Yusuff Dimeji.
2025. "Unraveling Drug-Induced Hepatotoxicity: Roles of Oxidative Stress, Biomarker Discovery, and Translational Advances" Trends in Applied Sciences Research 20, no. 1: 56-69. https://doi.org/10.3923/tasr.2025.56.69

This work is licensed under a Creative Commons Attribution 4.0 International License.